

Recent data from the U.S. Food and Drug Administration (FDA or the agency) on the percentage of De Novo classification requests that are declined or withdrawn raises questions about the prospects for De Novo submissions. Industry may need to temper expectations for a clear and uncomplicated standard to ease novel low- to moderate-risk devices to clearance. Careful planning and early engagement with the agency can enhance a company's understanding of FDA's requirements for particular devices.
FDA's De Novo statistics suggest a somewhat discouraging outlook for De Novo classification requests. The agency's Medical Device User Fee Amendments quarterly performance reports imply that getting through to clearance may be a highly variable undertaking, with relatively high rates of failure.
According to the most recent performance report, in 2018, there were 56 submissions for De Novo consideration, of which 31 (55%) failed in some way. In 2019, of 61 submissions, 20 to date have been declined or withdrawn (36 are still pending a decision).
Figure 1: CDRH - De Novo Performance Metrics - Rate of Grant, Decline, Withdrawal and Delete Decisions
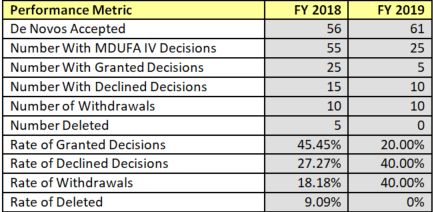
Although the agency's annual reporting metrics have changed since 2017, making direct comparisons inexact, the table below suggests that the recent data may be consistent with the post-2012 trend of numbers of initial requests that make it to a final grant of De Novo classification.1
Figure 2: De Novo Requests Disposition 2013 - 2017
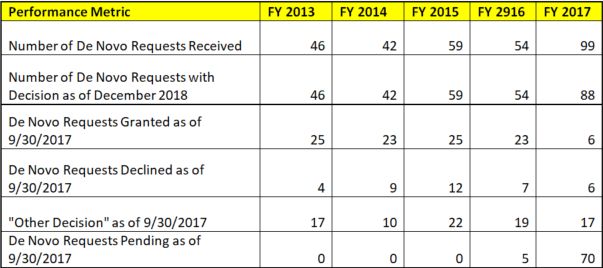
Data is based on the MDUFA III Performance Report (December 10, 2018) and cumulative summaries from prior year reports. After 2017, the MDUFA reports no longer present data on numbers of grants/declines from previous years' submissions.
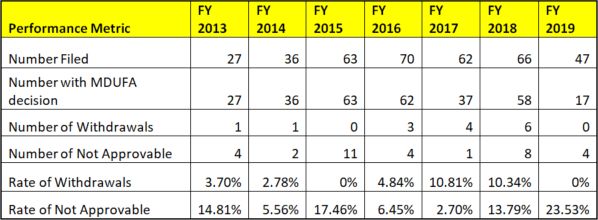
By contrast, data for original Premarket Authorizations (PMAs) indicate a much lower failure rate, with withdrawals/not approvables over the last seven years at approximately 12%.
Figure 3: PMA Original and Panel-Track Supplements (Without Panel Review), Withdrawal and Not Approvable Rates
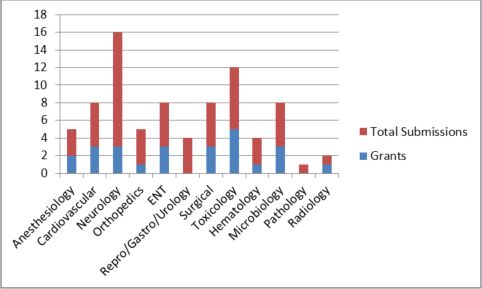
Variability in results for De Novo requests across divisions suggest that the challenges may differ significantly depending on product classification and review team. Likelihood of success appears to be especially low for neurology and reproductive/gastroenterology/urology requests.
Figure 4: De Novo Clearance Rate by Therapeutic Area – 2018
Should sponsors approach the De Novo process with caution?
It is not entirely clear what the reasons might be for the relatively low clearance rate of De Novo requests. One key driving factor may be inconsistency in applications or variability in threshold agreement as to how safe is safe and how effective is effective for novel, yet otherwise low- to moderate-risk devices. All companies are encouraged to use the pre-submission meeting process prior to filing a De Novo request to ensure better understanding of FDA's requirements prior to filing a De Novo request.
Footnote
1. In the 15 years following the creation of the De Novo classification process in the Food and Drug Administration Modernization Act (FDAMA) of 1997, there were only 76 total completed submissions. After the process was revamped in 2012 (FDASIA) to allow for direct submissions, annual numbers of De Novo classification requests have increased significantly.
The content of this article is intended to provide a general guide to the subject matter. Specialist advice should be sought about your specific circumstances.




