
Last week, the Medical Device Coordination Group (MDCG) published two new guidance documents under the Medical Devices Regulation (MDR) and In Vitro Diagnostic Devices Regulation (IVDR). These concern the "person responsible for regulatory compliance" and the "implant card" required under the new Medical Devices Regulations (MDR).
These are the latest of the guidance published by the MDCG and collated on the European Commission's website before the Regulations come into force in May 2020 (for medical devices) and May 2022 (for in vitro diagnostic medical devices).
Persons Responsible for Regulatory Compliance
Under Article 15 of the MDR and Article 15 of the IVDR, "Manufacturers shall have available within their organisation at least one person responsible for regulatory compliance who possesses the requisite expertise in the field of medical devices." While the Regulations set out requirements on the qualification of the PRRC and an overview of their responsibilities, the guidance adds additional detail to these requirements, and clarifies the PRRC requirements for manufacturers and authorised representatives (AR), notably that:
- Each legal manufacturer must have a PRRC at their disposal, and where organisations have more than one legal manufacturer within the group, each manufacturer must have their own PRRC. The guidance does not clarify if the same PRRC can act for multiple manufactures.
- Manufacturers that are not established in the EU must have an AR in a Member State of the EU, who must also have a PRRC. The AR may also subcontract the responsibilities of the PRRC to a third party. One individual cannot be the PRRC for both the manufacturer and the AR and, in the case of a micro or small enterprise, the PRRC of the manufacturer and the PRRC of the AR cannot be from the same external organisation.
- In terms of location, the PRRC should be located in the same place as the manufacturer or AR. Therefore, if a manufacturer is located outside the EU, the PRRC should also be located outside the EU. Manufacturers in the EU should likewise have a PRRC located inside the EU. As the AR will be located in the EU, the PRRC will also be located inside the EU.
- If the required qualifications of the PRRC were not obtained in the EU, they should be recognised by an EU Member State as equivalent to the EU corresponding qualification.
- Micro or small enterprise may subcontract the responsibilities of the PRRC to a third party, providing the PRRC has the relevant qualifications and the manufacturer can demonstrate they are able to fulfil their legal obligations while using a third party who is not within the manufacturers' organisation.
Note that the guidance does not refer to the potential liability of either the manufacturer or the PRRC for infringement of the PRRC's responsibilities, or on the relationship between the two entities. This may be addressed under national implementation of the MDR and IVDR. However, this lack of clarity will impact companies preparation for the MDR.
Implant Cards
Under Article 18 of the MDR, the manufacturer of an implantable device shall provide an implant card (IC) with the device, allowing the identification of the device, and other key information. In addition, Member States shall require healthcare institutions to make certain information available as part of the IC. Although the intended purpose and much of the required elements of the IC are included in the MDR, the IC guidance goes further, to include additional detail of the data elements, and provides examples of how this information should be presented.
In accordance with the MDR, the manufacturer must provide the following information with the IC:
- Device name;
- Serial number, lot number;
- Unique device identification (UDI);
- Name, address and the website of manufacturer;
- Device type.
The information should preferably be on the card itself, or as stickers to be placed on by clinicians, although this is discouraged in the guidance. The IC should also contain blank fields for the following information to be filled out by the healthcare provider:
- Name of the patient or patient ID;
- Name and address of the healthcare institution which performed the implantation;
- Date of implantation.
The dimensions of the IC should be credit card size. The guidance also advises on the use of symbols to avoid national versions of the IC, and contains a list of validated symbols that can be used for certain data fields. It recommends the use of an instruction leaflet to explain the symbols in the language of the relevant Member State. Finally, it suggests some designs for the implant card and the corresponding leaflet. The following is one of a number of examples provided in the guidance:
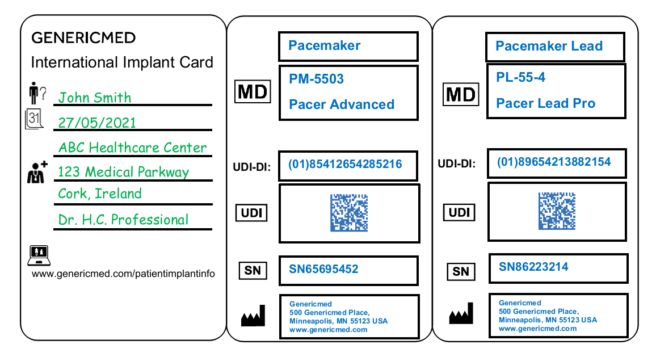
These practical examples will likely provide much needed clarity for companies, at least on this aspect of the MDR, and will be a useful tool to assist in the design and production of the ICs.
We would like to thank Julia Smith for her assistance with this blog post.
The content of this article is intended to provide a general guide to the subject matter. Specialist advice should be sought about your specific circumstances.
